

image from Freepik
General Knowledge
Structure of RBC
Red blood cells (RBCs) are the cells that carry oxygen from your lungs to the rest of your body and bring carbon dioxide back to your lungs to be exhaled.
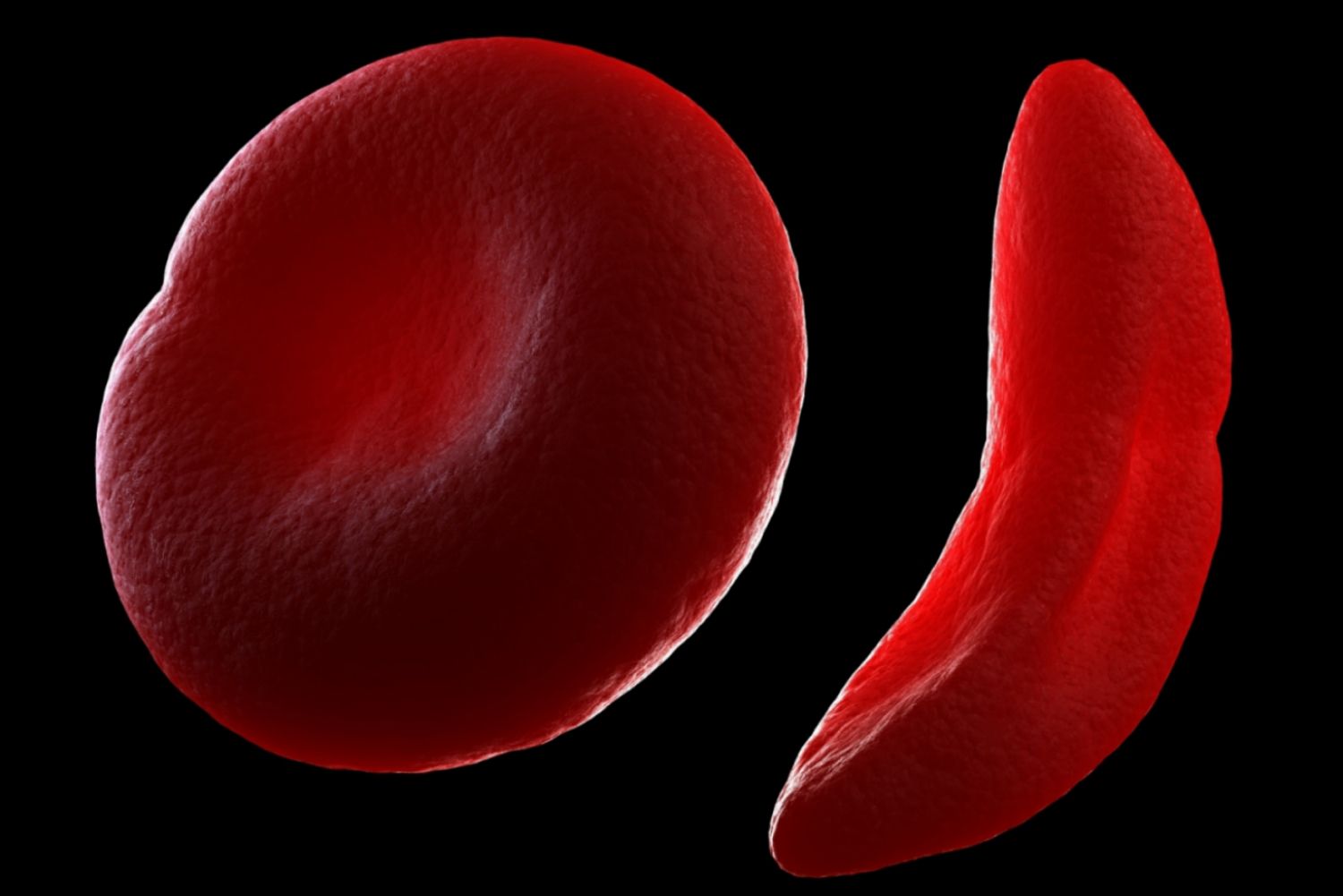
They are shaped like flexible discs, which helps them move easily through blood vessels. Their main component is hemoglobin, which is a special protein that actually carries oxygen.
Red blood cells live for about 3–4 months before the body breaks them down and makes new ones to keep the system running smoothly.
Main Idea: red blood cells are your body’s oxygen delivery system, constantly moving oxygen in and carbon dioxide out to keep you alive and energized.
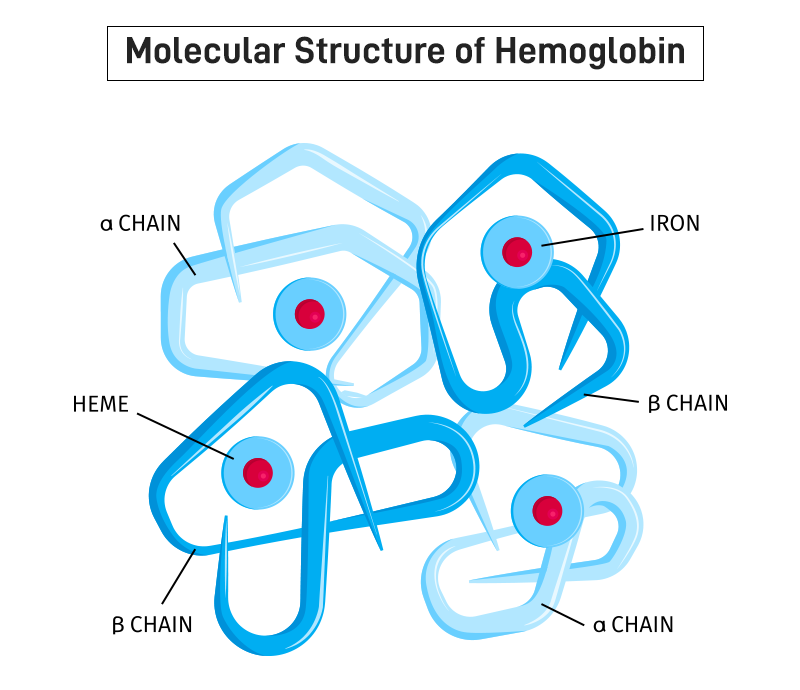
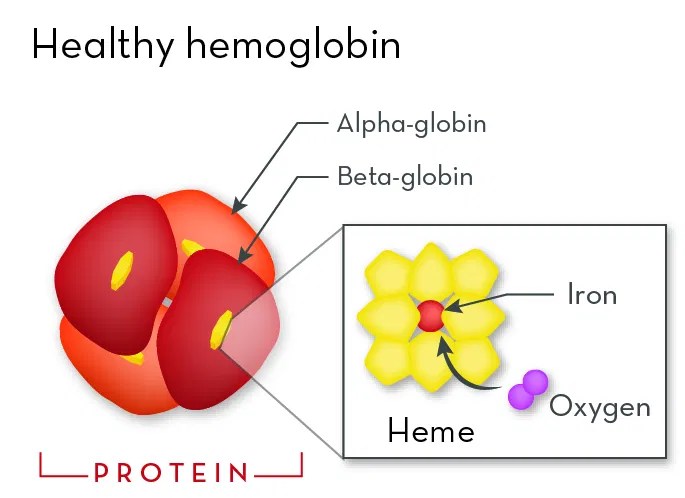
What is hemoglobin?
Hemoglobin is the protein inside red blood cells that carries oxygen. Normal adult hemoglobin, called HbA, is made of four protein chains: two alpha chains and two beta chains.
The beta chains are made according to instructions in the beta-globin gene, which is a part of your DNA that tells your body how to build this chain correctly.
When someone is “AA”, it means they have two normal copies of the beta-globin gene—one from each parent. This produces normal hemoglobin in all their red blood cells, which means that the cells stay round, flexible, and able to carry oxygen efficiently.
How did it start?
Sickle cell disease started as a genetic mutation in the beta-globin gene many generations ago. This mutation changes the shape of hemoglobin, producing a type called HbS instead of the normal HbA.
Sickle cell disease is most common in people whose ancestors come from parts of Africa, the Mediterranean, the Middle East, and India, because the sickle gene originally helped protect against malaria, which is a serious disease spread by mosquitoes. However, it can affect anyone if the genes are passed down.
Through history, people with one copy of the sickle gene were more likely to overcome malaria infections and survive and have children, thus pass that gene on to their children.

Why do sickle cells cause problems?
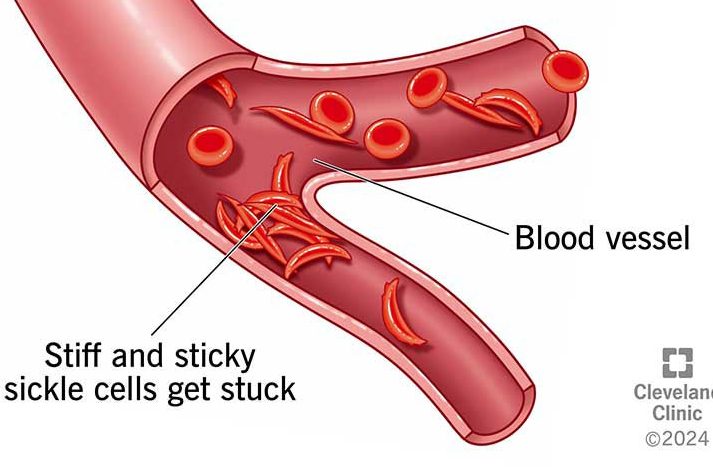
When a person has sickle cell disease, their red blood cells make a lot of HbS, the abnormal form of hemoglobin. Normally, hemoglobin carries oxygen without sticking together. But in HbS, the hemoglobin molecules can clump together, especially when oxygen levels in the blood drop, like during exercise or illness. This clumping changes the shape of the red blood cells, bending them from their normal round, flexible shape into a crescent or “sickle” shape.
These sickle-shaped cells are stiff and sticky. Because they can’t bend and flow smoothly like normal red blood cells, they often get stuck in small blood vessels. This can block blood flow, causing painful episodes, making organs work harder, and reducing the amount of oxygen that reaches the body.
Sickle cells also die much faster than normal red blood cells. While normal red blood cells last about 3–4 months, sickle cells only last 10–20 days. This rapid breakdown leads to a shortage of red blood cells, called anemia, which makes the body feel tired, weak, and short of energy.

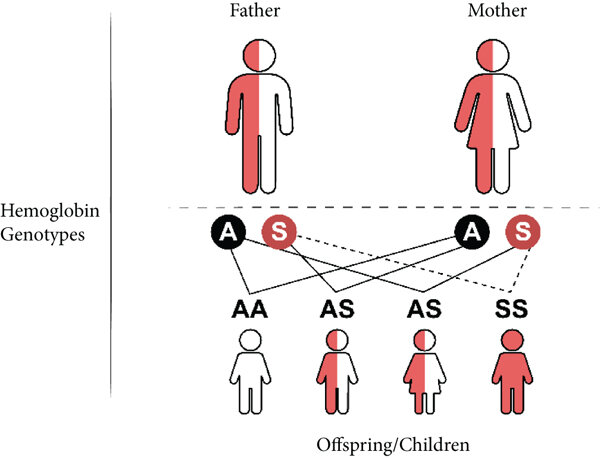
Hemoglobin Variants
Hemoglobin SC disease is a type of sickle cell disease that happens when a person inherits one sickle gene (S) from one parent and one hemoglobin C gene (C) from the other parent.
The hemoglobin C gene produces a slightly different abnormal hemoglobin than the sickle gene, and when both are present in the same person, their red blood cells can become partially rigid and abnormally shaped.
People with SC disease may experience anemia, pain crises, and complications such as joint swelling, organ damage, or vision problems, but these symptoms often occur less frequently or later in life compared with SS.
Those with AS, called sickle cell carriers, have one normal gene and one sickle gene. They usually do not have symptoms, but they can pass the sickle gene to their children. Similarly, people with AC have one normal gene and one hemoglobin C gene. Like AS carriers, AC individuals are generally healthy and symptom-free but can pass the C gene to their children.
Symptoms
One of the most common signs of sickle cell disease is anemia, which can make someone feel constantly tired, weak, or short of breath, even during everyday activities. You might notice pale skin or lips, because there aren’t enough healthy red blood cells carrying oxygen. Pain episodes, also called “crises,” are another hallmark symptom.

One of the most common signs of sickle cell disease is anemia, which can
These sudden episodes can cause sharp pain in the bones, joints, chest, or abdomen and can last hours or days. Swelling in the hands and feet is also common, especially in young children, and can be an early sign of blocked blood flow.
These symptoms can lead to more serious complications over time. For example, repeated pain crises and blocked blood vessels can cause organ damage, affecting the liver, kidneys, heart, and lungs. Strokes may occur if blood flow to the brain is blocked by sickle cells. Acute chest syndrome, a dangerous blockage in the lungs’ blood vessels, can cause severe chest pain, fever, and difficulty breathing.
Over time, these complications can make the body less able to cope with stress, illness, or infections, which is why careful monitoring, treatment, and education are crucial for people with sickle cell disease.

Treatments
image from Freepik
Sickle cell disease can be diagnosed shortly after birth through a simple blood test called a newborn screening, which checks for the presence of abnormal hemoglobin. The diagnosis may involve a blood test called a hemoglobin electrophoresis, which identifies the types of hemoglobin in the blood and confirms whether a person has HbA, HbS, or both.
Medications are an essential part of managing sickle cell disease. Hydroxyurea is a commonly prescribed drug that works by increasing the production of fetal hemoglobin (HbF), a type of hemoglobin that does not sickle. Higher levels of HbF help prevent red blood cells from forming the sickle shape, reduce pain crises, and lower the risk of complications such as acute chest syndrome.
Regular blood transfusions are sometimes recommended for people with severe sickle cell disease. Transfusions work by adding healthy red blood cells into the bloodstream, which helps increase oxygen delivery and reduce the number of sickle cells. This treatment can lower the risk of stroke in children, reduce severe anemia, and prevent complications in high-risk patients.
A bone marrow or stem cell transplant is currently the only treatment that can potentially cure sickle cell disease. In this procedure, a patient’s bone marrow, which produces red blood cells, is replaced with healthy marrow from a compatible donor. This allows the body to start producing normal hemoglobin and red blood cells. Transplants are most successful when performed in children, but they are complex procedures with significant risks, including infection or the body rejecting the donor marrow. Because of these risks, transplants are usually reserved for patients with severe disease or complications.
Treatments
FAQs
What is sickle cell disease?
Sickle cell disease is a genetic blood disorder caused by changes in the hemoglobin gene. It results in red blood cells that are abnormally shaped—rigid, sticky, and crescent-shaped—reducing their ability to carry oxygen and move smoothly through blood vessels. This can lead to anemia, pain episodes, and other health complications.
What does it mean to be a carrier?
A carrier has one normal beta-globin gene and one abnormal gene (either sickle or hemoglobin C). Carriers usually do not have symptoms but can pass the gene to their children. Common carrier types include AS (sickle cell trait) and AC (hemoglobin C trait).
Can sickle cell disease be cured?
Currently, bone marrow or stem cell transplants are the only potential cure, though they are complex and carry risks. Most treatments focus on managing symptoms and preventing complications. Emerging gene therapies are showing promise as a future cure.
How many people are affected in Canada?
An estimated 6,000 Canadians are living with sickle cell disease, and approximately 1 in every 2,500 newborns in Canada are affected by sickle cell disease.
Myth: Sickle cell only affects children.
Fact: Sickle cell disease affects people of all ages. While it is diagnosed in childhood, complications and symptoms continue into adulthood.
Myth: Sickle cell disease is contagious.
Fact: Sickle cell disease is genetic, not contagious. You cannot catch it from someone else.
Myth: Only people of African descent get sickle cell disease.
Fact: While it is most common in people with African ancestry, sickle cell disease can affect anyone whose parents carry abnormal hemoglobin genes.
Myth: People with sickle cell cannot live a normal life.
Fact: With proper medical care, monitoring, and lifestyle adjustments, people with sickle cell disease can live full, productive lives. Early treatment and preventive care greatly improve quality of life.
